

Los medicamentos similares en Uruguay tienen probada calidad farmacéutica ya que cumplen con la normativa vigente, y sin embargo no es posible asegurar que presentan los atributos necesarios para maximizar eficacia y seguridad ya que nunca fueron evaluados en seres humanos ni en ensayos que aporten evidencia respecto a su biodisponibilidad. Esto se explica por la evolución de la reglamentación uruguaya. Lograr que en Uruguay aumente la proporción de medicamentos bioequivalentes o intercambiables implica un avance en la regulación, y debe ser un objetivo común para todos los actores vinculados a la producción, gestión, prescripción, uso y regulación de medicamentos

Dr. QF. Manuel Ibarra
Prof. Agregado Biofarmacia y Terapéutica, CIENFAR, Facultad de Química. CEBIOBE. Udelar
Q.F. Yessica Imbriago
Estudiante de Posgrado en Química, Facultad de Química, Udelar.
Dr. Q.F. Marta Vázquez
Prof. Titular Biofarmacia y Terapéutica, CIENFAR, Facultad de Química. Directora del CEBIOBE. Udelar
Calidad Biofarmacéutica: un concepto necesario
La calidad farmacéutica es un concepto tan amplio como central en el ciclo de vida de los medicamentos. Las referencias a la calidad son continuas, y llamativamente encontrar una definición unánime no es tan sencillo. Podemos entender la calidad como “aptitud para el uso”, lo cual en el caso de los medicamentos nos conduce directamente a su rendimiento clínico. Bajo esta perspectiva, la calidad farmacéutica es un conjunto de atributos que confieren al medicamento una alta probabilidad de cumplir en forma reproducible con los objetivos de eficacia y seguridad cuando se utilizan en forma correcta en un paciente. A su vez, otra noción de calidad coexiste: la conformidad del producto con las normas establecidas en una determinada reglamentación. Estas concepciones no son necesariamente diferentes: la reglamentación puede estar diseñada de forma tal que maximice la aptitud para el uso de los medicamentos. Sin embargo, es importante notar que debido al peso que tienen las reglamentaciones en la actividad farmacéutica, el fin último puede quedar en un segundo plano.
El mercado farmacéutico uruguayo está compuesto en una alta proporción por medicamentos similares. Así se definen los productos que han sido desarrollados en base a un producto innovador u original, pero no han demostrado un equivalente rendimiento in vivo relativo a dichos productos. Nadie puede dudar que los medicamentos similares en Uruguay tienen probada calidad farmacéutica ya que cumplen con la normativa vigente, y sin embargo no es posible asegurar que presentan los atributos necesarios para maximizar eficacia y seguridad ya que nunca fueron evaluados en seres humanos ni en ensayos que aporten evidencia respecto a su biodisponibilidad. Esto se explica por la evolución de la reglamentación uruguaya. El patentamiento de productos farmacéuticos se hizo operativo a inicios del milenio. El requisito de bioequivalencia fue introducido para un listado inicial de principios activos en 2007 y luego ampliado en 2016. Medicamentos que contengan estos activos deben probar en un ensayo clínico concreto que poseen un atributo fundamental: su administración extravascular logra en seres humanos una exposición equivalente a la que alcanza el producto innovador, y por lo tanto habrá mayor probabilidad de que logre un rendimiento clínico también equivalente. Este esencial atributo de calidad puede definirse como calidad biofarmacéutica.
Lograr que en Uruguay aumente la proporción de medicamentos bioequivalentes o intercambiables implica un avance en la regulación, y debe ser un objetivo común para todos los actores vinculados a la producción, gestión, prescripción, uso y regulación de medicamentos. Avanzar hacia productos con calidad biofarmacéutica comprobada redundará en ventajas para los pacientes al reducir la variabilidad de la respuesta clínica asociada al producto, y en ventajas para el sector productivo ya que es un paso esencial hacia la innovación.
Screening in vitro/in silico: una alternativa para avanzar en Uruguay
La calidad biofarmacéutica debe ser evaluada in vivo, con la excepción de productos que contienen fármacos para los cuales se considera que se puede aportar suficiente evidencia obtenida in vitro para evitar la realización de un ensayo clínico (bioexención). Así lo entienden las agencias regulatorias de referencia mundial y cada vez más son los mercados que adoptan estas exigencias en su regulación.
Ahora bien, es necesario considerar las dificultades de avanzar en este camino en Uruguay, así como las particularidades de nuestro mercado farmacéutico: muchos medicamentos innovadores nunca se comercializaron aquí. Los medicamentos similares han dado disponibilidad de herramientas farmacológicas fundamentales para nuestra sociedad. En este contexto, exigir bioequivalencia para todos los principios activos en un corto o mediano plazo es impracticable, y probablemente innecesario.
Nuestro grupo de investigación se ha enfocado en incorporar metodologías novedosas para proyectar un camino que podría dar solución a la problemática actual. Metodologías que están provocando fuertes avances en el desarrollo de medicamentos innovadores. Brevemente, implica el uso de modelos farmacocinéticos basados en fisiología (PBPK) para el desarrollo de correlaciones in vitro-in silico-in vivo. Métodos que permiten predecir la calidad biofarmacéutica de una formulación incorporando la información obtenida en ensayos in vitro biorrelevantes dentro de modelos que incorporan conocimiento actual vinculado a la farmacocinética del fármaco en seres humanos, con el fin de reducir la necesidad de realizar ensayos clínicos y así alivianar costos económicos y éticos. Esto implica un avance respecto a las correlaciones in vitro-in vivo que sustentan la bioexención actualmente incorporada en nuestra regulación, ya que es aplicable a una significativamente mayor cantidad de principios activos.
La propuesta implica llevar adelante una línea proactiva de investigación en calidad biofarmacéutica de medicamentos similares, donde inicialmente se realice un screening mediante ensayos in vitro biorrelevantes acoplados a herramientas in silico para predecir el rendimiento biofarmacéutico. Esto permitiría detectar productos con potenciales inconvenientes, los cuales podrían ser subsecuentemente evaluados en estudios de bioequivalencia focalizados. Además, permitiría reforzar atributos de calidad biofarmacéutica de probablemente numerosos medicamentos similares sin la necesidad de conducir ensayos clínicos.
Prueba de concepto
Para aplicar este abordaje a la problemática, se han elegido medicamentos cuyo impacto socioeconómico sea marcado y/o cuyas formulaciones comerciales presentaran sensibilidad tal que pudieran implicar variabilidad en su rendimiento biofarmacéutico. Actualmente se desarrolla como prueba de concepto el proyecto
“Calidad biofarmacéutica de medicamentos similares conteniendo omeprazol evaluada mediante un enfoque In Vitro-In Silico-In Vivo” con financiamiento de la ANII.
El omeprazol es el inhibidor de la bomba de protones más ampliamente utilizado en alteraciones de la secreción acida gástrica, pero principalmente como gastro-protector.
Existen en Uruguay 11 medicamentos similares conteniendo omeprazol 20 mg. Debido a su inestabilidad en medio ácido (degradación significativa a pH<6) se comercializa en forma de cápsulas conteniendo microgránulos gastrorresistentes.
El omeprazol utilizado de forma crónica provoca un aumento en el pH gástrico basal hasta valores de 4-6, sin embargo, la capacidad del recubrimiento entérico se evalúa en farmacopeas oficiales a un pH cercano a 1. Este detalle muestra cómo los ensayos farmacopeicos no necesariamente se diseñan para evaluar el rendimiento in vivo. La biodisponibilidad de omeprazol tras dosis múltiples estará fuertemente afectada por el rendimiento del recubrimiento entérico de cada formulación en medio gástrico a valores de pH en los cuales no es evaluado.
Se desarrollaron entonces ensayos in vitro para caracterizar el rendimiento de las formulaciones en condiciones biorrelevantes, considerando dos características fundamentales: el pH al cual se comienza a disolver el principio activo y el comportamiento relativo entre formulaciones considerando una etapa ácida (gástrica) a pH 4 seguida de una etapa casi neutra (intestinal) a pH 6.8.
Vale destacar que el omeprazol, como la gran mayoría de fármacos, se absorbe esencialmente en intestino delgado. Cuanto más tiempo se encuentre retenido y disuelto en estómago a pH<6, más se degrada y por lo tanto pierde biodisponibilidad. En intestino si bien puede disolverse a valores de pH entre 4 y 6, la alta permeabilidad del activo hace que rápidamente se absorba y no de tiempo a una significativa degradación a nivel luminal. Para estudiar la interacción de estas variables (zona de disolución, velocidad de degradación y velocidad de absorción), los resultados obtenidos in vitro se incorporan en modelos PBPK para predecir el rendimiento final in vivo y así detectar productos con potenciales diferencias importantes en biodisponibilidad. Finalmente, estos productos se evaluarán in vivo mediante un estudio de bioequivalencia en dosis única y dosis múltiples, que aportará insumos fundamentales para validar la correlación in vitro-in silico-in vivo desarrollada y así extrapolar los resultados a formulaciones no evaluadas en el ensayo clínico.
Resultados parciales del proyecto: rendimiento de las formulaciones en condiciones in vitro biorrelevantes
Con el fin de caracterizar el rendimiento in vitro de las 11 marcas de cápsulas de omeprazol 20mg comercializadas a nivel nacional, se realizaron dos tipos de estudios: 1) evaluación de la resistencia del recubrimiento entérico a pH ácido creciente; y 2) evaluación de los perfiles de disolución simulando dosis única (DU), con una etapa ácida a pH 1,2 y simulando dosis múltiple (DM), donde la etapa ácida se realizó a pH 4. En ambos casos la etapa ácida fue proseguida de un cambio de medio para obtener el perfil de disolución a pH 6,8. Para los productos con rendimiento más disímil, se realizó, además, un ensayo in vitro adicional evaluando la sensibilidad para la cantidad final de omeprazol disuelta en base a la duración de la etapa ácida a pH 4, con el fin de evidenciar cambios en la performance de las formulaciones ante tiempos de vaciamiento gástrico diferentes. Estos ensayos fueron complementados con estudios de estabilidad de omeprazol en distintas condiciones y ensayos de disolución en distintos tipos de aparato. Se muestran en este artículo sólo algunos de los resultados obtenidos hasta el momento.
Los resultados muestran importantes diferencias de rendimiento entre las formulaciones. En cuanto a la resistencia del recubrimiento entérico, se observó que un único producto no liberó principio activo en el rango de pH evaluado (de 3 a 5), mientras que se encontraron cinco formulaciones que liberaron más del 20% de la cantidad total de OMP al finalizar el ensayo en pH 5. En cuanto a la evaluación de los perfiles de disolución, se encontraron diferencias significativas tanto en la velocidad de disolución como en la cantidad disuelta a tiempo final. A su vez, en concordancia con los resultados obtenidos en las pruebas de resistencia de la cubierta entérica, varios productos mostraron diferencias de rendimiento entre las condiciones DU y DM, evidenciando que la pérdida del recubrimiento entérico a pH probables de encontrarse tras 5 días de tratamiento provoca una disminución significativa en la cantidad de omeprazol disponible para ser absorbida. En la figura 1 se presentan los perfiles de liberación del omeprazol desde todas las formulaciones en el rango de pH creciente ensayado.
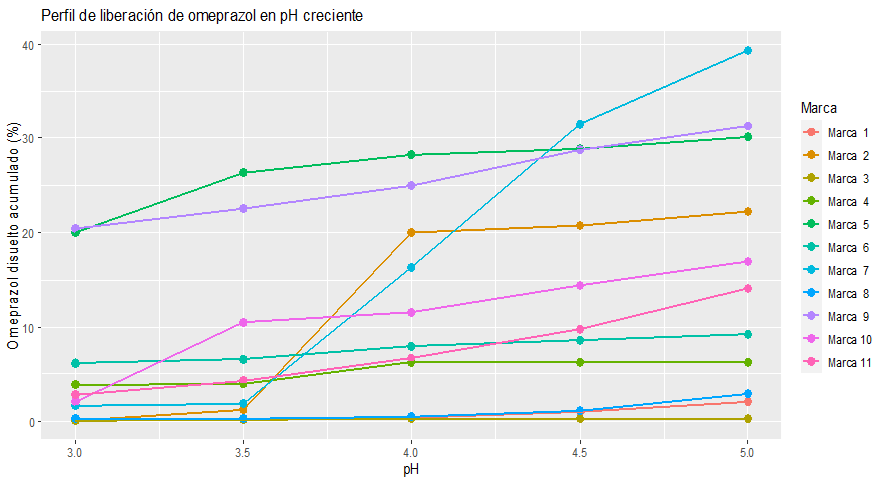
Figura 1. Perfil de liberación de omeprazol para cada formulación en el rango de pH creciente evaluado. Obtenidos en aparato IV USP – celda de flujo continuo.
El ensayo de sensibilidad ante duración de la etapa ácida en condiciones de DM realizado a las dos formulaciones con comportamiento más diferente entre sí puso de manifiesto que la variabilidad en el tiempo de vaciamiento gástrico y por tanto el tiempo de exposición de la formulación a un pH ácido, dan como resultado una diferencia significativa en la cantidad de omeprazol que finalmente queda disponible para ser absorbida desde el intestino. Como puede observarse en la Figura 2, la marca A no libera OMP en la etapa ácida y por lo tanto alcanza una similar cantidad disuelta a pH 6,8 en ambos ensayos. Sin embargo, la marca B libera OMP a pH 4, por lo que la cantidad total disuelta de OMP a tiempo final es más de 2 veces mayor cuando la etapa ácida es más corta.

Figura 2. Perfiles de disolución de las marcas con comportamiento más disímil, en la prueba de sensibilidad por duración de etapa ácida en condiciones de DM (pH 4).
Los resultados encontrados permiten proyectar diferencias significativas en la calidad biofarmacéutica de las marcas comerciales disponibles en Uruguay. A la vez, se evidencia que para ciertos productos la biodisponibilidad podría disminuir tras dosis múltiple.
Proyecciones
El proyecto financiado por ANII incluye la realización de un estudio de bioequivalencia a dosis única y dosis múltiple entre las formulaciones que presentaron los rendimientos in vitro e in silico más disímiles. Este estudio se llevará adelante este año, y sus resultados permitirán evaluar la precisión y exactitud de la predicción in silico realizada, con lo que se podrán proyectar los rendimientos in vivo de todas las formulaciones evaluadas in vitro en este proyecto.
Se espera que los resultados de este estudio sirvan para concientizar a actores clave sobre el impacto de la variable calidad biofarmacéutica entre medicamentos similares, y fundamentalmente que consoliden herramientas que pueden aportar soluciones para avanzar hacia la disponibilidad de medicamentos seguros y eficaces y hacia la innovación farmacéutica en Uruguay.

Nota
El presente proyecto con financiación de la Agencia Nacional de Investigación e Innovación (ANII – FMV_3_2018_1_147996) se desarrolla hasta el momento en el Centro de Evaluación de Biodisponibilidad y Bioequivalencia de Medicamentos (CEBIOBE) de la Udelar. Participan como supervisoras analíticas y bioestadísticas la Dra. Marianela Lorier y la Q.F. Alejandra Schiavo, y han contribuido en los ensayos in vitro numerosos estudiantes en el marco de practicantados y trabajos experimentales, dentro del plan de formación de recursos humanos del CEBIOBE. Asimismo, integran el equipo del proyecto que avanzará a la fase clínica: Dr. Pietro Fagiolino (FQ-Udelar), Dr. Iñaki F. Trocóniz (U. Navarra – España), Bach. Andrés Baptista, Q.F. Natalia Guevara (FQ-Udelar), Dra. Cecilia Maldonado (FQ-Udelar), Dr. Nicolás González (FMed – Udelar), Dr. Alejandro Goyret (CEBIOBE y FMed-UdelaR) y el Laboratorio All Quimia Ltda.